It is impossible to imagine the medical device industry today without the software revolution. From defibrillators to infusion pumps and robotic surgical systems, a broad range of devices relies on software to function safely and effectively. At the same time, medical device software has introduced a level of complexity that dwarfs anything seen before in the field. This column addresses some basic facts about medical device software and how quality professionals, together with executive management, can work together to ensure that FDA’s rigorous requirements are satisfied.
Note that FDA has separate requirements for medical device software and quality system software. This column addresses only the software that is part of a medical device, such as software that triggers an alarm when a product fails. Other software, such as that used by a medical device manufacturer to manage complaints in its quality system, is outside of the scope of this discussion.
In October 2017, FDA released two new final guidance documents on the same day: “Deciding When to Submit a 510(k) for a Change to an Existing Device” and “Deciding When to Submit a 510(k) for a Software Change to an Existing Device.” In other words, medical device software changes have so many unique challenges and risks that they earn a guidance document of their own, separate from all other device changes.
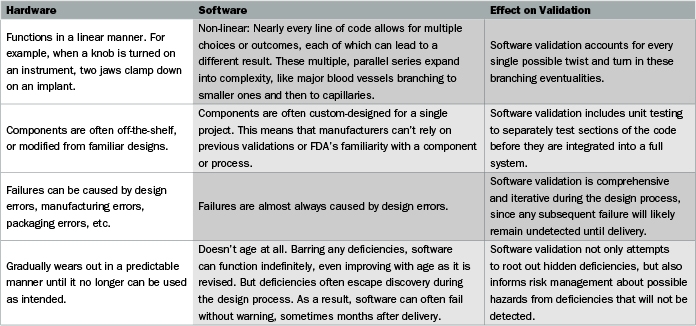
The Problem: Software Is Different from Hardware
Validation is at the heart of device design, and the validation of software design is especially challenging. Even though FDA’s guidance document, “General Principles of Software Validation,” was last updated in January 2002, that guidance is still highly relevant and useful. When a medical device incorporates software, FDA expects the manufacturer to be well-read in the guidance document.
Furthermore, FDA does not intend the guidance document on software validation to be read only by software developers or quality engineers, as it states, “Software engineering needs an even greater level of managerial scrutiny and control than hardware engineering.” The guidance is written in laymen’s terms, so executive management is not excused from this responsibility, even when they have limited experience in software development.
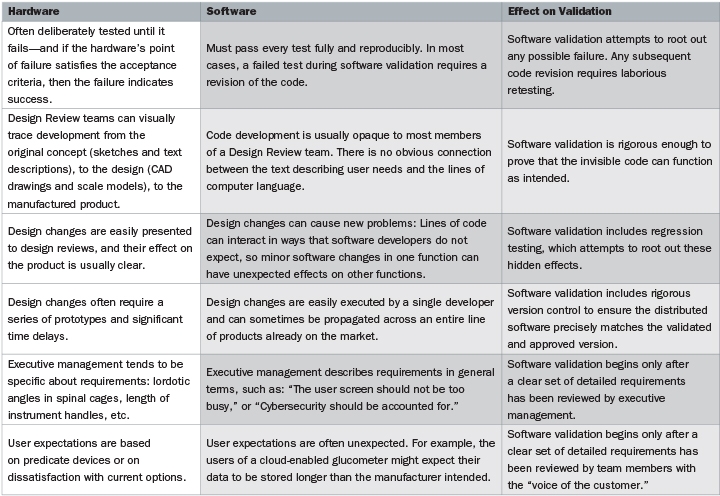
To get to the heart of the problem, the guidance document on software validation includes the deceptively simple statement: “Software is different from hardware.” Actually, there are many complex differences between software and hardware, and understanding those differences is key to ensuring that software validation will pass FDA muster. The comparison chart (above and on the previous page) is adapted from, and expands on, the FDA guidance.
The Solution: Software Validation Driven by Rigorous Requirements
Both of the last two differences in the chart use the phrase, “a clear set of detailed requirements.” This is the most crucial element for proper software validation, and one that is frequently neglected. The flow chart figure on page 20 illustrates how requirements play an early and crucial role in software development.
During the phases that developers are coding and testing the software, the requirements enter a tunnel that is closed to non-developers, and the software emerges from the other side as a complete design. The resulting software can support a safe and effective device only if executive management and other stakeholders have reviewed a detailed and unambiguous set of requirements. Quality and regulatory teams can expedite this crucial phase by ensuring smooth communications between engineering and the rest of the organization.

At the far end of the tunnel, quality and regulatory conduct the final stages of user site testing with faithful attention to the original requirements. As noted in the list of differences between software and hardware, “user expectations are often unexpected,” and any expectations that were not properly specified as requirements are likely to emerge as errors during testing.
Clearly, the future of medical device development is bound up with new advances in software—wearable devices, remote medicine, algorithmic diagnostics, and robotics. FDA expects that manufacturers’ quality systems and design controls will ensure safety and efficacy, even as the software code at the heart of the device remains opaque to executive management. Software design might be more challenging than hardware, but software validation will keep the differences manageable and the quality undiminished.
Dan Goldstein is a manager for Quality Assurance at Musculoskeletal Clinical Regulatory Advisors (MCRA), primarily focusing on quality system requirements for bringing new devices to market and keeping experienced manufacturers in compliance with FDA and Notified Bodies. He provides MCRA clients with gap assessments, mock FDA inspections, Form 483 remediations, Design History Files, Technical Files, Summary Technical Documents, and Clinical Evaluation Reports. A graduate of the University of Maryland University College, Dan has worked since 2002 in quality assurance for medical devices, including autologous blood products for wound healing and computer-aided-detection software for lung diseases. Musculoskeletal Clinical Regulatory Advisers LLC has broad experience in the area of software validation. MCRA’s staff is especially adept at promoting and maintaining the lines of communication that keep executive management, the “voice of the customer,” and software developers on the same page with regard to the detailed requirements that drive the development process. The organization believes in requirements that follow the “four Cs”—clear, concise, correct, and complete.